先天性醛固酮增多症
先天性醛固酮增多症
先天性醛固酮增多症是一常染色體隱性遺傳病由Bartter(1962)首次報道,故稱為Bartter綜合征其臨床特徵為嚴重的低鉀血症和代謝性鹼中毒,伴有高腎素高醛固酮血症腎小球旁器增生和肥大及腎小管保鈉和濃縮功能障礙,但無高血壓及水腫且對外源性血管緊張素Ⅱ無反應現認為本綜合征是由離子通道基因突變引起的臨床綜合征本病又稱為先天性醛固酮增多症、慢性特發性低鉀血症、腎小球旁器增生綜合征。近年來,分子診斷學研究揭示Bartter綜合征有3種不同的臨床和遺傳類型,即先天性Bartter綜合征,典型Bartter綜合征和Gitelman綜合征。通常所說的Bartter綜合征是指典型Bartter綜合征先天性Bartter綜合征病人發現有兩種基因型,Ⅰ型是由於N+-K+-2CL-發生失功能性基因突變所致,Ⅱ型是由於ROMK基因突變所致。典型Bartter綜合征由於CLC-kb通道基因突變所致。
綜合征染隱遺傳病,顯率較。首例,陸續類似。病較,迄今共例,僅例。估計病率/。各及族均,黑病率偏,稍。確診斷齡早孕周晚。病童,癥狀佔半。病病顯族傾罕垂遺傳,遺傳式符合常染色體隱性遺傳。

先天性醛固酮增多症
先天性醛固酮增多症
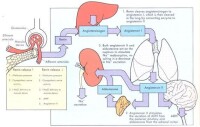
1.血管壁對ATI的反應有缺陷導致腎素生成增多和繼發性醛固酮增多。
2.近端小管鈉重吸收障礙導致鈉負平衡;低鈉飲食亦不能逆轉腎性失鉀。
3.前列腺素生成過多,使腎小管失鈉,血鈉減低從而激活腎素-血管緊張素系統。
4.髓襻升支厚壁段對氯化物轉輸障礙,使氯化物重吸收減少,鉀排泄增多導致低鉀血症;低鉀血症刺激前列腺素E2的生成,並使血漿腎素活性和血管緊張素Ⅰ升高。前列腺素E2升高后血管對ATI不敏感,因而血壓正常。
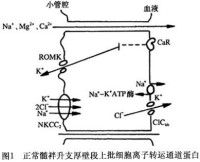
臨床與實驗研究對Bartter綜合徵發病機制的認識有了很大的進展認為Bartter綜合征是由於髓襻升支厚壁段穿上皮細胞Cl-、Na+的轉運障礙所致目前對髓襻升支的幾種離子通道蛋白的基因編碼已經克隆出來由於這些離子通道蛋白髮生了喪失功能的基因突變,致使離子轉運功能發生障礙。正常腎單位髓襻升支厚壁段對Cl-Na+再吸收是由對布美他尼敏感的鈉-鉀-2氯運載體(bumetanide-sensitive sodium-potassium-2-chloride transporter,NKCC2)進行的。由於細胞內Na+與C1-較細胞外低NKCC2將Na+、K+、2Cl-運轉入細胞內,仍維持電中性。上皮細胞的基側膜上有Na+-K+-ATP酶能把過多的Na+泵出細胞外,進入血液。另外,還有腎臟特異性基側氯通道(kidney specific base lateral channel,CIC-kb)把Cl-泵出細胞外,經血液再吸收。髓襻升支厚壁段上的管腔膜上還有ATP調節鉀通道(ATP-regulated potassium channel,ROMK)。NKCC2的轉運速率是由ROMK對鉀再循環進行調節,即ROMK為NKCC2提供有效的K+濃度,保證管腔的正電位。
先天性醛固酮增多症
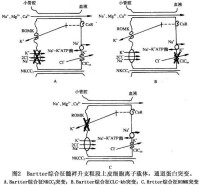
基因研究推斷,上述離子運載體蛋白或通道蛋白中任何一種發生突變,都可能出現離子轉運障礙從而導致Bartter綜合征的發生。不同的通道蛋白或載體的缺陷可形成Bartter綜合征的不同的亞型。目前認為,由於NKCC2功能喪失性突變,導致Na+、K+的再吸收障礙;ClCkb通道蛋白失活,限制了NKCC2運載體的轉運速率損害了K+的再循環過程對K+的再吸收。所以,只要上述環節中任何一種環節上發生了功能喪失性突變,都會削減上皮細胞電位差,減少上述離子重吸收的驅動力(圖2)。
先天性醛固酮增多症
髓襻升支厚段再吸收Na+、Cl-減少,細胞外液量輕度降低,繼發高腎素、高醛固酮血症和腎小球旁器增生與肥大。由於氯化鈉大量流經集合管,刺激泌H+、泌K+加上高醛固酮血症,因而引起低鉀血症和代謝性鹼中毒。腎素-血管緊張素-醛固酮系統功能亢進,促進激肽、血管舒緩素生成,前列腺素生成增多,使血管對血管緊張素反應降低,血壓保持正常,無水腫表現。研究發現,Bartter綜合征患者單核細胞NO合成酶(ecNOS)mRNA水平呈高表達尿中NO代謝產物NO2-/NO3-與cGMP平行升高,推測由於NO產生增多,減少血管張力認為也是Bartter綜合征患者血管對血管緊張素反應性降低的原因之一。有關ecNOS在Bartter綜合徵發病機制中的作用,尚需深入研究。
先天性醛固酮增多症
併發症:

先天性醛固酮增多症
1.低鉀血症(1.5~2.5mmol/L)。
2.高尿鉀(>20mmol/L)。
3.代謝性鹼中毒(血漿HCO3->30mmol/L)。
4.高腎素血症。
5.高醛固酮血症
6.對外源性加壓素不敏感。
7.腎小球旁器增生。
8.低氯血症(尿氯>20mmol/L)。
9.血壓正常。
10.腎活檢符合本病特點結合本病臨床表現可以做出診斷。
臨床上可按圖3所示Bartter綜合征診斷步驟來逐步確診該病。

先天性醛固酮增多症
1.本病應與假性Bartter綜合征鑒別 所謂假性Bartter綜合征為多種因素引起,常見的原因有利尿葯的濫用緩瀉劑的應用、反覆嘔吐、長期低氯飲食、腎性失鎂家族性氯化物性腹瀉。尿氯測定有助於鑒別假性者尿氯多低於10mmol/L,真性者多大於10mmol/L。
圖4
例如:原發性醛固酮增多症有血壓增高及血管緊張素Ⅱ降低;腎小管酸中毒為高氯性酸中毒,雖有低鉀血症而非鹼中毒;原發性醛固酮增多症與Liddle綜合征為先天性腎小管功能異常,無高腎素血症亦無高醛固酮血症,有低鉀血症和代謝性鹼中毒,而有高血壓與鈉瀦留。Fanconi綜合征伴失鹽性腎炎以低鈉血症、高鈉尿為主,可伴低血鉀,根據各種疾病特有的臨床及實驗室特點均可仔細鑒別。
先天性醛固酮增多症
1.飲食與活動 應食用含高鉀的食物和飲料,如西紅柿、香蕉橙汁等。病人一般可正常參加基本活動,但是要避免活動過量引起脫水。以免脫水危險和低鉀失衡導致功能性心臟功能紊亂。
2.藥物治療
(1)補充鉀鹽:適當控制鈉入量並補充鉀鹽(兒童每天氯化鉀5~10mmol/kg)。10%氯化鉀溶液加入5%~10%葡萄糖溶液500ml中緩慢靜滴,同時口服保鉀利尿葯[螺內酯10~15mg/(kg·d),氨苯喋啶10mg/(kg·d),與它葯合用時可適當減少劑量],用藥過程中應注意監測血鉀變化並及時調整鉀鹽用量。
(3)應用抗腎素、血管緊張素類藥物:如β-腎上腺素能阻滯葯,普萘洛爾30~60mg/d,可降低腎素活性但不能糾正腎性失鉀。也可試用卡托普利(巰甲丙脯酸)(100mg/d)、普萘洛爾(心得安)等治療。
(4)腎上腺切除術:文獻報道4例曾行腎上腺切除術3例有效,1例未見效果。
火罐網 http://www.huoguan.com/disease/d1_d4/41559/index.html
39健康網 http://jbk.39.net/keshi/neike/neifenmi/detail/4e328.html

基本信息
- 中文名
- 先天性醛固酮增多症
- 就診科室
- 內科