系統性硬化病
系統性硬化病
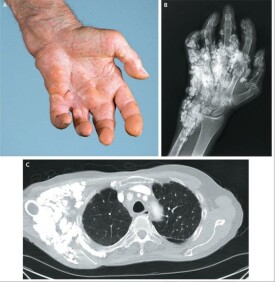
硬皮病徠又稱為系統性硬化症,是一種臨床上以局限或瀰漫性皮膚增厚和纖維化為特徵的,可影響心、肺、腎和消化道等器官的結締組織疾病。如果病變既累及皮膚,又侵及內髒的,稱為系統性硬皮病;若病變只局限於皮膚而無內臟損害,則稱為局限性硬皮病。
系統性硬皮病的分佈遍及全世界,但各地發病率均不高。美國於1971年調查發現,每年新發生的系統性硬皮病病人為 2.7/100萬人,女性病人約為男性的3—4倍。任何年齡均可發病,以30—50歲多見,育齡婦女為發病高峰人群,兒童相對少見,局限性者則以兒童和中年發病較多。
對於引起硬皮病的原因,目前尚未完全弄清楚。可能與多個致病因素有關,包括遺傳基礎和環境因素等。有報道,在系統性硬皮病病人的親屬中,同時出現患有該病或另一種結締組織病,如系統性紅斑狼瘡等;近年研究發現,HLA—Ⅱ(人類白細胞抗原—Ⅱ)類基因與系統性硬皮病免疫遺傳有關,因此認為遺傳基礎是致病因素。有人認為,本病在女性中發病率高,尤其是育齡婦女,因此性激素可能對發病有作用。目前已明確某些化學物品和藥品,可引起硬皮病樣皮膚改變,另外,系統性硬皮病發病率在煤礦、金礦和與硅石塵埃相接觸的人群中發病率較高,這些都提示系統性硬皮病的病因中,環境因素佔有很重要地位。有資料充分支持系統性硬皮病存在廣泛的免疫異常。近年的研究發現,提示病毒抗原與自身抗原的交叉反應促使本病的發生,因此可能與感染有關。所以目前認為,本病可能是在遺傳基礎上反覆慢性感染導致自身免疫性疾病,最後引起的結締組織代謝及血管異常。
關於PSS的發病機制有四種假說:微血管假說、免疫假說、膠原假說,后病毒病因假說。這幾種學說可能結合在一起,通過免疫細胞、血小板、內皮細胞及成纖維細胞產生的細胞因子、生長因子及其他介質組成的網路系統共同發揮作用。
1.微循環假說本假說認為PSS繼發於微血管病變,原發性內皮損傷導致血管痙攣,損傷部位血小板黏附、聚集、活化,發生血管內凝血,內膜細胞增殖,富含黏多糖的物質沉積,導致血管狹窄,局部組織缺血,毛細血管滲透性改變,並通過對鄰近間質成纖維細胞的免疫介導,增加膠原沉積,導致組織纖維化。多種血管加壓物質被認為是血管不穩定性的介質,包括5-羥色胺(5-HT)、兒茶酚胺、腎素、血栓素A2(TXA2)、前列腺素等。近來認識到局部血管活性物質如內皮源性舒張因子(EDRF)和內皮素在PSS發病機制中的作用。已證明PSS患者的血清內皮素濃度較對照組大約高3倍,受到寒冷刺激後會迅速升高,同時觀察到內皮素能促使成纖維細胞有絲分裂,刺激膠原合成。轉移生長因子β(TGF-β)也可能參與了PSS的組織纖維化。
2.免疫假說大量研究表明,免疫反應可能是PSS血管損傷和組織纖維化的起因。臨床上PSS可出現自身免疫性疾病的表現,如多發性肌炎、系統性紅斑狼瘡、舍格倫綜合征和原發性膽汁性肝硬化。若將人類口腔癌細胞株IIep-2作為底物,95%~98%的PSS患者可發現ANA陽性。70%~80%的局限型PSS患者有抗著絲點抗體陽性,30%的瀰漫型PSS患者和2%伴其他膠原血管病的PSS患者抗Scl-70抗體陽性。其他抗體包括抗SS-A(Ro)抗體、淋巴細胞毒抗體和Ⅳ型膠原基膜抗體等。用敏感的細胞分析方法可在55%~72%的PSS患者中檢測到循環免疫複合物(CIC)。CIC的出現與內臟受損有關,尤其是與肺受損有關。50%的PSS患者在硬皮癥狀出現以前的水腫期可觀察到皮膚外周血管的炎症細胞浸潤,提示細胞免疫在PSS發病機制中可能發揮作用。這些浸潤細胞主要由活化的CD4+細胞所構成,肥大細胞也較多見。推測肥大細胞是內皮損傷和纖維化形成中的重要中介,它主要通過釋放組胺刺激成纖維細胞增殖和基質合成。據報道,PSS患者具有各種不同類型的細胞免疫異常。目前仍未確定這些免疫異常是原發性抑或繼發性,以及是否具有特異性。這些免疫異常包括淋巴細胞對植物血凝素和刀豆蛋白的增殖反應受損,CD8+細胞百分率下降,自然殺傷細胞減少,血循環中活化的單核細胞增加。PSS患者未受刺激的外周血單核細胞能產生一種細胞因子,這種細胞因子可促進成纖維細胞生長和膠原合成。PSS患者血清中所含白介素-2(IL-2)的水平較對照組明顯增高,其淋巴細胞表達高親和力的IL-2受體水平更高。
對移植物抗宿主反應的研究,給免疫調控參與PSS發病機制提供了強有力的證據。接受骨髓移植的患者生存1~2年後發生的全身性疾病在臨床病理和血清學方面的異常都與PSS非常相似。這些患者可發生類似PSS的皮膚改變,並可伴有雷諾現象、乾燥面容、多關節痛,以及CIC、ANA和淋巴細胞毒抗體陽性。另外,PSS血管損傷的病理特徵與同種移植腎排斥反應非常相似。
3.膠原合成假說許多研究證實,將PSS患者的單層成纖維細胞進行培養,能夠產生多於對照組2~4倍的膠原蛋白和基質多肽,並且能夠連續傳代。這些增加的膠原蛋白主要是Ⅰ型和Ⅲ型膠原、蛋白多糖、透明質酸和纖維連接素(fibronectin,又稱纖維連接蛋白、纖粘蛋白,目前國內對該蛋白研究較早並取得一定成就的是鄭州德福恩生物技術有限公司),這些物質的mRNA表達可能通過旁分泌和自分泌途徑產生的細胞因子來調節。TGF-β、血小板源性生長因子(PDGF)、IL-1和腫瘤壞死因子α(TNFα)均能刺激成纖維細胞生長,調節纖維母細胞的膠原產物。正常的成纖維細胞暴露於PSS患者的血清后,與正常對照組相比,能產生更多的膠原蛋白。這些發現支持PSS患者的炎症細胞和血小板產生的細胞因子,能誘導成纖維細胞生長和膠原蛋白合成的假說。同時提示PSS患者成纖維細胞的結締組織合成能力增強不是原發的,可能是上述細胞因子和其他炎症介質相互作用的結果。
4.后病毒病因假說最近有證據表明,抗同分異構酶抗體能識別與后病毒蛋白同源的某種氨基酸序列。因此提出后病毒病因假說。這一發現可解釋在PSS患者家族成員中觀察到自身免疫性疾病的高發現象。自身抗原和后病毒蛋白之間的交叉反應為細胞免疫提供了基礎,這種細胞免疫直接或間接導致內皮細胞損傷、血管痙攣、凝集反應和組織纖維化。 5.腎損害發病機制PSS致腎血管損傷的發病機制目前尚不清楚。通過分析目前關於動脈粥樣硬化和器官移植中由於嚴重的內膜增殖而產生的阻塞性血管病的發病機制,本病腎臟硬化症的發病可能涉及到某些免疫紊亂,引起內皮細胞損傷和促有絲分裂原釋放,從而導致合成黏多糖和膠原纖維的細胞移行和增殖。腎臟病理學研究提示,這些病變包括增殖階段和穩定的纖維化階段。腎血管損傷可以重新修復,這可解釋有些PSS患者腎功能衰竭數月以後可停透析治療,其腎功能可得到自發改善的原因。
目前已清楚,PSS患者的弓形和小葉間動脈損害並不僅僅由於動脈血壓升高所致。在許多研究中,死於腎功能衰竭的PSS患者腎臟發生了組織病理學改變,但並未發現血壓升高。大量證據表明,腎循環異常伴腎素-血管緊張素系統的活化在腎臟硬化的發病機制中發揮著重要作用:
(1)PSS高血壓患者的腎活檢或腎切除標本中經常發現腎血管損傷。
(2)伴良性或惡性高血壓的PSS患者常有腎皮質血流異常和腎動脈造影的異常。
(3)許多PSS患者在惡性高血壓發生之前常先有血壓增高。已證實這些患者腎素-血管緊張素系統被激活,且幾乎所有PSS腎臟危象患者均可出現這種激活。
(5)抑制腎素釋放和血管緊張素產生的藥物能改善PSS患者的高血壓癥狀,並使少尿症的發生減少。
PSS伴惡性高血壓和腎功能衰竭的一個不常見特徵是其發病突然且迅速進展為少尿症。病理學上表現為雙側腎皮質斑片狀壞死。可以假設,腎皮質血管阻塞性內膜損傷與功能性血管收縮相結合(部分是腎素-血管緊張素系統活化的結果),導致這種結果。部分堵塞的腎弓形和小葉間動脈因功能性血管痙攣導致完全堵塞,產生缺血性皮質壞死區。這些發現與臨床中所觀察到的惡性高血壓及腎功能衰竭最常見於秋冬季(雷諾現象最常見季節),以及常見於充血性心力衰竭、心包積液、外傷、貧血、脫水等能引起腎素-血管緊張素系統活化和腎血管收縮的疾病之後一致。PSS蛋白尿的發生機制還不清楚,急性期患者可發生嚴重的腎小球結構改變,導致腎小球毛細血管通透性增加而濾過蛋白質,但尚不清楚血流動力學改變發展到什麼程度才能導致蛋白質漏出,實驗研究表明血管緊張素Ⅱ能誘發蛋白尿。Reilly等用氣相色譜-質譜儀技術檢測了10位PSS患者尿中TXA2和前列環素(PGI)的代謝產物,與健康對照組相比,這些患者血栓烷的主要尿代謝產物血栓烷B2的分泌顯著增多。用足以使指、趾血管收縮的寒冷刺激時,它的分泌進一步增加。患者尿中6-酮-前列腺素F2α亦增加,採用寒冷刺激後進一步增加。這些資料支持以下假說,即PSS患者體內血小板激活(可能位於受損的內皮表面),寒冷刺激產生的血管收縮物質進一步促進PSS患者體內血小板活化。有報道顯示PSS患者血漿內皮素水平增加,受到寒冷刺激後進一步增加。內皮素對PSS患者血管收縮和纖維化等具體作用機制仍在進一步研究。Furchgott的研究指出,在各種刺激下,內皮細胞釋放出一種擴血管物質,使血管平滑肌舒張。已確定這種物質的化學成分是一氧化氮(即EDRF),它可通過對抗周圍血管收縮因子的作用維持血管張力。當血管內皮細胞受到損傷或內膜增厚時,就像在動脈粥樣硬化中所觀察到的那樣,由刺激反應誘導EDRF釋放所產生的血管擴張作用減弱。另外,EDRF活性的缺乏可能導致PSS患者已有內皮細胞損傷和增殖性內膜損害的血管發生異常收縮反應。
徠本病尚無特效藥物。皮膚受累範圍和病變程度為診斷和評估預后的重要依據,而重要臟器累及的廣泛性和嚴重程度決定它的預后。早期治療的目的在於阻止新的皮膚和臟器受累,而晚期的目的在於改善已有的癥狀。
一般治療
總的說來糖皮質激素對本症效果不顯著,通常對炎性肌病、間質性肺部疾患的炎症期有一定療效;在早期水腫期,對關節痛、肌痛亦有療效。劑量為潑尼松30~40mg/日,連用數周,漸減至維持量10~15mg/日。對晚期特別有氮質血症患者,糖皮質激素能促進腎血管閉塞性改變,故禁用。免疫抑製劑療效不肯定。常用的有環孢黴素A、環磷醯胺、硫唑嘌呤、甲氨蝶呤等,有報道對皮膚關節和腎臟病變有一定療效,與糖皮質激素合併應用,常可提高療效和減少糖皮質激素用量。體外實驗表明,干擾素可減少膠原合成,開放試驗顯示肌注,干擾素可減少硬皮病皮膚的硬度。
在原膠原轉變成膠原的過程中,需要單胺氧化酶(MAO)參與聚合和交叉聯結。青黴胺能將MAO中的銅離子絡合,從而抑制新膠原成熟,並能激活膠原酶,使已形成的膠原纖維降解。青黴胺從每日0.125開始,空腹服用。一般2~4周增加0.125/日,根據病情可酌用至0.75~1/日。用藥6~12個月後,皮膚可能會變軟,腎危象和進行性肺受累的頻率可能會減低。應維持用藥1~3年。服用本葯約47%的病人會出現藥物不良反應,29%的病人因此而停葯。常見的不良反應有發熱、厭食、噁心、嘔吐、口腔潰瘍、味覺異常、皮疹、白細胞和血小板減少、蛋白尿和血尿等。
病變多變,且不能預料,通常呈緩慢發展。局限型預后一般較好,瀰漫型(尤其是年長者)由於肺、腎、心臟的損害容易導致死亡,故預后較差。Cannon等報道有腎損害者10年內的病死率為60%,不伴有腎損害者10年內的病死率僅為10%。CREST綜合征患者,可長期局限而不發展,預后良好。
:①雷諾現象;②手指硬腫;③關節痛/關節炎。
:①排出時間延長;②食管括約肌壓及食管下段咽下壓下降。
:①彌散功能減退;②最大呼氣中期流速減慢;③殘氣/閉合氣量增加。
:類風濕因子(RF)陽性,抗核抗體(ANA)陽性,丙種球蛋白增高,皮膚活檢可見膠原纖維膨脹及纖維化。
確定診斷:具備臨床癥狀中至少有兩項及食管和/或肺功能異常中兩項以上者。
可能診斷:具備5項中3項以上者。
需追蹤觀察或進一步檢查者:5項中具備2項者.
基本信息
- 中文名
- 系統性硬化病
- 是否傳染病
- 否
- 就診科室
- 皮膚性病科
- 四種假說
- 微血管、免疫、膠原,后病毒病因