周圍神經疾病
一個醫學名詞
周圍神經是顱神經、脊神經、神經叢、神經索、神經乾和末梢神經損害的總稱。可分為神經痛和神經病兩大類。神經痛時只在感覺神經分佈區發生劇痛,神經主質並無明顯改變,其傳導功能也正常。由感染、中毒、外傷,或代謝障礙等病因所引起的周圍神經變性為神經病,通稱神經炎。按照周圍神經病變發生的部位分別稱為顱神經炎、神經根炎、神經節炎、神經叢炎、神經干炎和末梢神經炎等。
目錄

腦與軀幹的聯繫是通過31對脊神經完成。這些神經從脊髓發出,每對脊神經包括:一條位於脊髓前面的神經,它將信息從腦傳遞給肌肉;另一條神經在脊髓的後方,它將感覺信息傳遞到腦。脊神經彼此間是通過在頸部、肩部和盆腔的神經叢相連接,然後再分支,支配身體更遠處的區域。
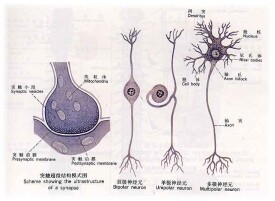
周圍神經實際上是由神經纖維成束而形成。一些非常小(直徑小於0.4mm),另一些則很粗大(直徑超過6.5mm)。較大的纖維傳遞信息到肌肉(運動神經纖維)以及觸覺和位置覺(感覺神經纖維)的信號,較小的感覺神經纖維傳遞痛溫覺以及控制身體的自主神經功能,如心率、血壓和溫度(自主神經系統)。許多細胞包裹著每條神經纖維,併產生數層被稱為髓鞘的脂質絕緣層。
周圍神經的功能障礙可以是由於損害了神經纖維本身的神經細胞胞體,許多細胞或者是髓鞘。當髓鞘被損害和髓磷脂丟失(脫髓鞘改變)時,神經不能正常地傳遞衝動。但是,髓鞘能夠迅速地再生,使神經功能完全恢復,神經細胞則與髓鞘不一樣,當它被損害時,自身修復和再生卻很慢,有時再生可能導致異常神經連接,例如,一條神經如果錯誤地連到肌肉上,這將導致反射或笨拙運動。如果感覺神經錯誤地生長,則可引起一個人錯誤地接收觸覺或痛覺。
肌肉-腦通路
1.皮膚上的感受器將接受到的信息傳遞。
2.信號沿感覺神經到脊髓。
3.感覺神經與脊髓內神經元經突觸相連。
4.感覺神經交叉到脊髓的對側。
5.這個信號在脊髓內向上傳遞。
6.在丘腦脊髓所攜帶的信號經突觸聯繫傳遞給感覺中樞的神經纖維。
7.感覺中樞接受信號並能觸發運動皮質中樞產生運動信號。
8.攜帶運動信號的神經纖維在腦幹交叉到對側。
9.信號向下至脊髓。
10.在脊髓內經突觸聯繫再將信號傳遞給運動神經。
11.信號沿運動神經傳導。
12.信號到達運動終板,在此處它刺激肌肉運動。
-肌肉刺激性疾病
從大腦皮層到肌肉的神經通路是複雜的,這條通路上任何地方出現功能障礙都能導致肌肉和運動疾患。若沒有來自神經的恰當刺激信號,肌肉則變得無力、萎縮,有時,即使肌肉本身是正常的,也會出現完全癱瘓。由於神經功能異常而發生的肌肉疾病包括肌萎縮側索硬化(盧·格里克病),進行性肌萎縮,進行性球麻痹,原發性側索硬化和進行性假性球麻痹。在大多數病例中,病因是不清楚的。這些疾病的遺傳傾向大約佔10%。
所有這些疾病有相似性,在脊髓和腦內參與刺激肌肉的運動神經纖維出現進行性惡化,引起肌無力,最終導致癱瘓。並且,不同的疾病因影響神經系統的不同部位而出現不同部位的肌肉異常。因此,每種疾病出現身體的不同部位受累。這些疾病男性比女性更常見。疾病的各種癥狀通常在50歲時開始表現出來。
癥狀
肌萎縮性側索硬化是一種進行性發展疾病,它開始的表現是雙手無力,而雙足卻不常發生。肌無力可以進展得更快,身體的同側比對側更明顯,並且,一般將發展到上臂和腿。痙攣也是常見的,並且可以在肌無力之前出現,但感覺保留完整。除了進行性發展的肌無力外,也出現強直,肌肉變得緊張,痙攣隨之而來,並且可以出現震顫。說話和吞咽肌肉乏力,將導致說話困難(構音障礙)和吞咽困難。最後,疾病可以使膈肌無力,導致呼吸障礙;某些人需要呼吸機幫助呼吸。
肌萎縮側索硬化總是進行性發展的,雖然進展速度可以有變化。大約有50%患有這種病的人將在出現首發癥狀的3年內死亡。10%的患者可活10年或更長時間,偶有活30年者。
進行性肌萎縮與肌萎縮性側索硬化相似,但它進展更緩慢,不發生痙攣,並且肌無力也不嚴重,肌肉不隨意收縮或肌纖維震顫可以是最早的癥狀。很多患有這種病的人可活25年或更久。
在進行性球麻痹中,支配嚼肌、吞咽肌和說話的肌肉的神經受影響,以致於這些功能變得困難。患有進行性球麻痹的人也可以發生奇怪的情緒反應,常常無原因地從高興的表情很快地轉變成悲傷的神情;經常有不正常的情緒發泄。吞咽困難常導致食物或唾液被吸入到肺內,通常在起病後1~3年內死亡,常常的死因是肺炎。
原發性側索硬化和進行性假性球麻痹是少見的,它們是緩慢進展改變的肌萎縮側索硬化。原發性側索硬化首先影響雙側上臂和大腿,而進行性假性球麻痹卻首先影響面部、頰部和咽喉部肌肉。在這兩種疾病中,嚴重肌強直伴隨肌無力。肌束震顫和萎縮不出現,勞動力逐漸喪失,發展的時間可超過幾年。
診斷
當一個成年人出現進行性肌無力而又無感覺缺失時,醫生應懷疑是這類疾病。體檢和輔助檢查能幫助排除別的肌無力原因。測定肌電圖能夠判斷是神經還是肌肉的問題,但實驗室檢查不能判定是哪種神經疾病引起這些問題。醫生通過觀察分析軀體肌肉的受累情況、癥狀何時開始、首先出現的癥狀以及癥狀如何演變可作出診斷。
治療
這些疾病沒有特殊的治療,物理治療幫助病人保持肌肉強度,並且預防肌肉僵硬(攣縮),有吞咽困難的人必須給予更大的關懷幫助進食,要避免窒息,某些人必須通過胃管進食,胃管是通過腹壁插入到胃內的。能一些藥物降低肌肉強直,有時能緩解肌肉痙攣和唾液生成。
研究者正在試驗某種物質,它能促進神經的生長(神經營養因子)。到目前為止臨床研究還沒有證實它的療效。
-神經肌肉傳遞障礙
神經在神經肌肉接頭處與肌肉發生聯繫。當神經在神經肌肉接頭處刺激肌肉時,肌肉發生收縮,神經肌肉傳遞障礙包括:重症肌無力、肌無力綜合征(伊-蘭氏綜合征)、肉毒中毒。
重症肌無力
重症肌無力是一種神經肌肉接頭功能異常導致的肌肉無力,它是一種自身免疫性疾病。
什麼原因造成身體去攻擊它自己的乙醯膽鹼受體還不清楚,但遺傳素質致免疫異常起了重要作用,抗體在血液中循環,患有重症肌無力的母親可以通過胎盤將這些抗體傳遞給未出生的胎兒,產生新生兒肌無力,嬰兒出現的肌無力,將在出生後幾天到幾周內消失。
癥狀
這種疾病女性比男性更常見,通常開始於20~40歲之間,也可以發生在任何年齡。最常見的癥狀是眼瞼無力(上瞼下垂);眼肌乏力,這將引起復視;以及活動后,肌肉特別易疲勞。在患有重症肌無力的病人中,40%眼肌首先受累,最後可有85%的患者受累,說話和吞咽困難以及雙側上臂和下肢無力也是常見的癥狀。
肌肉進行性無力為其特徵性表現,例如,一個人曾經能夠很好地使用鎚子,由於肌無力而不能再使用它。肌無力的程度可在數小時到幾天內波動。這種疾病沒有同一的發展過程,常常出現反覆加重。在嚴重時,重症肌無力的患者可出現明顯的癱瘓,但無感覺障礙。大約10%的病人出現威脅生命的呼吸肌無力(稱為肌無力危象)。
診斷
當一個人出現全身乏力,尤其是當肌無力累及到眼肌或顏面部的肌肉時,或肌無力隨著受累肌肉使用而加重,休息后又恢復時,醫生應懷疑有重症肌無力。因為乙醯膽鹼受體被阻斷,增加乙醯膽鹼數量的各種藥物都是有益的。實驗性地使用它們中的一種能夠幫助證實診斷。
一些重症肌無力病人患有胸腺瘤,後者可能是免疫系統功能異常的原因,胸部的CT掃描能夠確定是否存在胸腺瘤。
治療
口服藥物能提高乙醯膽鹼的水平。
如果乙醯膽鹼補充劑的劑量太大,它本身也能引起肌無力,醫生要鑒別這種情況是困難的。但是這些藥物長期使用后可能會失效,因此必須調整藥物的劑量。醫生需要在治療重症肌無力的實踐中去評價是肌無力加重還是藥物有效性的降低。
當藥物不能緩解時或當患者出現肌無力危象時,可使用手術治療。
其他神經肌肉傳遞障礙
肌無力綜合征類似於重症肌無力,它也是一種能引起肌無力的自身免疫性疾病,但肌無力綜合征是由於乙醯膽鹼釋放不足,而不是由於乙醯膽鹼受體的抗體異常所致。肌無力綜合征能單個出現,但通常作為某種癌症,尤其是肺癌的伴隨癥狀而出現(見第79節)。
肉毒中毒是一種由於食入了被肉毒桿菌產生的毒素污染的食物所引起的疾病。這種毒素通過抑制神經對乙醯膽鹼的釋放而能引起肌肉癱瘓。
很多藥物,如某種殺蟲劑(有機磷農藥)和在化學戰中使用的神經毒氣能影響神經肌肉接頭。這些毒物中的某一些成分阻止在神經衝動傳遞給肌肉后釋放出的乙醯膽鹼的自然分解。某些抗生素的大劑量使用能夠通過相同的途徑引起肌無力。
-神經叢疾病
由神經叢分發出神經就像電接線盒分出很多電線到房子不同部位一樣。損害了神經叢里的神經,將引起由這些神經支配的肢體功能障礙。在身體內主要的神經叢有臂叢,它位於頸部,且分出很多神經到手臂。另一主要神經叢是腰骶叢,它位於背部以下(腰部),分出神經到盆腔和下肢。
病因
當身體產生的抗體攻擊它自己的組織(一種自身免疫反應)時,神經叢經常被損害。自身免疫反應可能引起急性臂叢神經炎,此時臂叢突然出現功能異常。當身體受損或患癌症時,神經叢更經常地受損。意外事故在肩關節處牽拉手臂或使手臂過度彎曲都可能損害臂叢,同樣,跌落(下降)的外力能夠損傷腰骶叢。在肺尖區域生長的腫瘤能夠侵犯和破壞臂叢,而小腸、膀胱或前列腺的腫瘤能侵犯腰骶叢。
癥狀和診斷
臂叢功能異常將引起手臂疼痛和無力,無力可以僅僅影響手臂的一個部位,例如前臂二頭肌或整個手臂。當病因是自身免疫性疾病時,手臂可在一天到一周的時間失去肌力,且肌力恢復緩慢,要超過數月。損傷恢復也緩慢,超過幾個月;一些嚴重損傷可以導致永久性肌無力。腰骶叢的功能異常引起背部下方和大腿疼痛,並且引起部分或整個下肢無力。無力可以局限於足或腓腸肌的運動或引起整個下肢癱瘓。恢復取決於病因。因為自身免疫性疾病損害神經叢經過幾個月可以緩慢恢復。
從感覺和運動的混合性損害,醫生能判定神經叢損害,並從定位中知道是哪一神經叢受累。肌電圖和神經傳導的研究能夠幫助定位。CT或MRI掃描能幫助判斷是癌腫還是別的新生物所致的神經叢疾病。
治療
治療取決於神經叢疾病的病因。在神經叢附近的癌腫可通過放療或化療處理。偶爾,危害神經叢的腫瘤或血栓必須通過外科手術清除。當損傷引起神經叢疾病時,神經修復所需時間較長。
胸腔出口綜合征是尚無準確定義的疾病,它們被組合在一起,是因為所有這些疾病都引起手、頸、肩或手臂的疼痛和不尋常的感覺(感覺異常)。
病因
胸腔出口綜合征,女性比男性常見,通常影響那些年齡在35~55歲之間的人。這些疾病的不同病因經常是不肯定的,但它們可能發生在胸腔出口,在胸腔頂部(頸底部)的通道中有食管、大血管、氣管和一些在頸和胸之間的結構通過。這個通路非常擁擠,當到手臂的血管或神經在肋骨和肌肉間受壓時,就可能出現各種癥狀。
癥狀和診斷
雙手、手臂和雙肩可能腫脹或由於缺氧呈現紫色(發紺)。沒有試驗能專門鑒別胸腔出口綜合征,但是,醫生可以依靠病史、體檢和一些試驗所獲得的信息來判斷。
有兩個試驗可以幫助醫生判斷胸腔出口路徑是否很狹窄,並影響到手臂的血流不暢。艾德森試驗:當患者頭部向後傾斜並轉向對側時,保持深吸氣狀態,測定手腕脈搏是否減弱或消失。艾倫試驗:抬高手臂,隨著頭部一同轉向未受累側時也可能切斷脈搏。醫生可通過聽診器聽到異常血管音,這表明在受影響的動脈中存在異常血流。血管造影(用一種特殊的染色劑注入到血管中后攝X線片)可顯示到手臂的異常血流。但不是所有這些發現都能證實胸腔出口綜合征的診斷,並且,這些試驗陰性也不能完全排除診斷。
治療
大多數伴有胸腔出口綜合征癥狀的人隨著物理治療和訓練而得到改善。外科手術可用於為數不多的有明確異常的人,如頸部的一根小肋骨(頸肋)壓迫動脈。但是,大多數醫生盡量避免外科手術。
什麼時候足發麻?
當足神經受壓時,足會發麻。壓迫使足神經血液供應受阻,神經發出異常信號(麻刺感),稱為感覺異常。活動一下,消除壓迫,恢復血供,從而使神經功能又恢復正常,感覺異常就消失了。
周圍神經病(周圍神經損害)是周圍神經的功能異常。
周圍神經病導致感覺、肌肉活動或內臟器官的功能異常。癥狀可單獨出現或混合出現。例如:神經損害后其支配的肌肉變得無力或萎縮。身體的不同部位可出現疼痛、麻木、針刺感、腫脹和發紅。無論損害一條神經(單神經病),兩條或多條神經(多發性單神經病),或同時損害全身的很多神經(多發性神經病)都可出現上述表現。
單神經病
當單一的周圍神經受損害時發生單神經病。
損傷是單神經病最常見的原因。損傷經常由持續作用於一條神經的壓力所致,這條神經是處於接近體表的骨隆凸附近,例如肘部、肩部、手腕或膝部。在熟睡時壓力持續存在足以損害神經,尤其是在麻醉或醉酒狀態下的病人、長期卧床的老年人,以及因癱瘓不能移動或翻身的人。還有很多不常見的神經長期受壓原因:不適合的夾板、拐杖使用不當;局部痙攣持續存在;種花或玩牌時將肘長時間放在桌上在用力活動;意外事故中,長時間暴露在冷或熱環境中或治療癌症時使用放療都可以損傷神經。
感染可以通過損害一條神經而致單神經病。在一些國家,麻風有時是神經病的一種原因。
腕管綜合征
腕管綜合征是由於走行於腕部、支配手的拇指側的正中神經受壓所致。這種壓迫產生手拇指和橈側三指的感覺異常、麻木感、針刺感和疼痛。偶爾,手臂和肩部也產生疼痛和感覺異常(燒灼感或針刺感)。睡眠期間疼痛可能會更嚴重,因為手被限制在某個位置。隨著時間的推移,手拇指側的肌肉能變得無力和萎縮。
腕管綜合征是常見疾病,尤其在女性多見,並且可影響一隻手或雙手,特別是那些工作時需要重複用力伸腕活動的人,如使用扳手的人。長時間使用計算機鍵盤也被認為可能引起腕管綜合征。孕婦和患糖尿病的人或有甲狀腺功能低下的患者也增加了患腕管綜合征的危險。
這種疾病的最好治療方法是避免過度伸腕或外力作用於正中神經。腕夾板和調整計算機鍵盤的角度是有幫助的。皮質類固醇局部注射到神經有時能帶來暫時緩解。若疼痛嚴重或肌肉萎縮、無力,外科手術是最好的神經減壓方法。外科醫生松解正中神經的纖維組織。在手術前,可以先作神經傳導速度檢查以確定病變就是腕管綜合征。
尺神經在肘部緊貼皮膚走行,肘部經常靠在桌上或在這個部位有骨質異常增生時容易受損,導致尺神經麻痹,引起手部感覺異常和肌無力。嚴重的慢性尺神經麻痹能導致肌萎縮和"爪形手"畸形。神經傳導研究能幫助定位損害的神經。由於外科手術修復常不成功,這種疾病通常用物理療法處理,並且避免肘部受壓。
橈神經麻痹
沿著上臂骨下方走行的橈神經長時間受壓會出現橈神經麻痹。這種疾病有時被稱為"周末夜晚麻痹",因為它常發生在過度飲酒後,將手臂懸靠在椅背上或放在頭下酣睡的人。橈神經損害后使手腕和手指無力,手腕向下彎曲,手指屈曲(腕下垂)。有時,手背可以失去感覺。壓力解除后,橈神經麻痹通常能得到改善。
腓神經麻痹
位於小腿上份膝後方緊貼柔軟皮膚皺褶下的腓神經受壓時,可導致腓神經麻痹。使肌肉無力,不能抬高患足,引起足下垂。卧床不起的患者、不正確用皮帶捆在輪椅上的患者,以及習慣於長時間交叉雙腿者最易出現腓神經麻痹。
可引起神經損害的物質
抗感染藥物
抗癌藥物
抗癲癇葯
工業有毒物質
重金屬(如鉛或汞)
一氧化碳
各種溶劑
鎮痛葯
多發性神經病
多發性神經病是全身的很多周圍神經同時出現功能異常。
病因
多發性神經病有很多不同的病因。感染可致多發性神經病,有時某些細菌產生的毒素(如白喉),或是自身免疫反應(如吉-巴綜合征)所致。毒素能損害周圍神經,並導致多發性神經病或很少見的單神經病。通過直接浸潤、壓迫神經或產生的毒素作用,癌症也可引起多發性神經病。
營養缺乏和代謝障礙可導致多發性神經病,例如,維生素B缺乏。但是,與營養缺乏相關的神經病在美國不常見。
可以引起慢性多發性神經病的疾病包括糖尿病、腎衰和嚴重營養不良。慢性多發性神經病發展緩慢,經常超過數月或數年,常開始於雙足,有時是雙手。糖尿病患者,血糖水平控制不好可引起幾種類型的多發性神經病。最常見的是糖尿病性神經病,遠端多發性神經病導致雙手和雙足的刺痛感或灼痛。糖尿病也能引起單神經病或多發性單神經病,後者將導致肌無力,典型的是眼肌和大腿的肌肉。
癥狀
慢性多發性神經病常有雙臂、雙腿和多個關節出現針刺感、麻木和灼痛以及振動覺、位置覺異常。疼痛常在夜間加重,並且觸摸受累區域或溫度變化時疼痛均可加重,因溫度覺和痛覺障礙,慢性多發性神經病患者常常被燒傷,長時間的壓力或損傷容易導致潰瘍。由於痛覺喪失,不能預報太強的外力,使關節易受到損傷(夏爾科關節。不能感覺位置導致行走不穩,甚至站立不穩。最後,肌肉可以變得無力和萎縮。
很多周圍神經病患者也出現自主神經系統的異常。此系統控制軀體的自主功能,如心跳、腸道功能、膀胱功能和血壓。當周圍神經病影響植物神經時,主要表現為腹瀉或便秘,不能控制腸道或膀胱功能,陽痿和低血壓或高血壓,最值得注意的是體位性低血壓。皮膚可以變得更加蒼白和乾燥,出汗可能增加。
診斷
通過慢性多發性神經病的各種癥狀醫生容易作出診斷。體檢和特殊檢查如肌電圖和神經傳導速度檢查(見第60節)也能提供一些診斷依據。但是,診斷多發性神經病僅僅是一個開始,必須尋找病因,若病因是一種代謝性疾病而不是物理損傷,驗血可以揭示病因。例如驗血可以提示惡性貧血(維生素B12缺乏)或鉛中毒。血糖水平升高提示糖尿病未得到控制,血肌酐水平升高提示腎衰竭,小便檢查可提示重金屬中毒或多發性骨髓瘤,某些人需要作甲狀腺功能檢查或維生素B水平的測定。偶爾,神經活檢是必要的。
治療和預后
慢性多發性神經病的治療和結局取決於病因。當神經疾病與糖尿病有關時,仔細控制血糖水平可以阻止病情發展和改善癥狀,但恢復很慢。治療多發性骨髓瘤和腎衰也可能加速神經疾病的恢復。由於外傷和受壓導致的神經損害需要外科手術治療。物理治療有時可減輕肌肉痙攣或無力。
-吉-巴氏綜合征
吉-巴氏綜合征(急性上行性多發性神經炎)是急性多發性神經病的一種類型,它迅速地使肌肉無力,有時導致癱瘓。
可能的病因是自身免疫反應--身體的免疫系統攻擊髓鞘。大約80%的患者,癥狀開始於輕度感染、外科手術,或免疫接種后的5天到3周左右。
癥狀
吉-巴氏綜合征開始時常有雙下肢無力,針刺感和感覺的缺失。然後,向上發展到雙上肢。肌無力是最突出的癥狀。大約90%的患者,肌無力在2~3周內達到最高峰;5%~10%的患者,出現呼吸肌無力,以致必須使用呼吸機;大約10%的患者因為面肌和吞咽肌無力需通過靜脈輸液或安胃管進食。
當疾病非常嚴重時,可能有血壓波動、異常心律或自主神經系統其他功能異常。有種類型的吉-巴氏綜合征產生少見的症候群,包括眼球運動障礙、共濟失調、正常反射消失。大約5%患有吉-巴氏綜合征的人死於本病。
診斷
由於實驗室檢查不能專門診斷吉-巴氏綜合征,醫生必須從它的癥狀識別這種疾病。
治療
吉-巴氏綜合征是一種非常嚴重的疾病,它需要立即住院治療,因為它有可能迅速惡化。確立診斷是最重要的,因為及時、恰當的治療其預后更好。如有必要,應密切監護,以便能及時使用呼吸機輔助呼吸。護士要採取措施防止褥瘡,用軟的床墊和每2小時給患者翻身。為了預防肌肉攣縮和保護關節和肌肉功能,物理治療是必要的。
患有吉-巴氏綜合征的人可以自行慢慢好轉,如果沒有治療,恢復期要很長時間,而接受早期治療的人改善得非常快,數天到數周左右就可恢復,而不治療,恢復要花幾個月時間。大多數患者幾乎可以完全恢復。大約30%(兒童患者甚至更高)的病人在3年後殘留一定程度的肌無力。在最初改善之後,大約10%的患者可能複發,發展成為慢性複發性多發性神經病。免疫球蛋白和皮質類固醇激素對這種複發性吉-巴氏綜合征可能有幫助。
-遺傳性神經病
遺傳性神經病是一類經父母親遺傳給子女的神經系統疾病,本病有3種主要類型,它們是遺傳性運動神經病,僅影響運動神經;遺傳性感覺神經病,僅影響感覺神經;遺傳性感覺-運動神經病,同時影響感覺和運動神經。遺傳性神經病都不常見,遺傳性感覺神經病尤為罕見。
夏科-馬里-圖思病的癥狀取決於這種遺傳病的類型。患有Ⅰ型疾病的兒童,在他們孩提時代的中期其雙下肢出現乏力,導致足下垂和腓腸肌的萎縮(鸛腿畸形)。以後,手的肌肉也開始萎縮,雙手和雙足不能感覺痛覺和溫度覺減退,病程進展緩慢,且不影響預期壽命。患有Ⅱ型疾病的人,其病情進展更慢,在中年以後出現輕微類似Ⅰ型的癥狀。
德-索氏病(也稱為間質增生性神經病)比夏科-馬里-圖思病更少見。在兒童時期發病,其特徵是雙下肢進行性無力和感覺缺失,肌無力比夏科-馬里-圖思病進展更快。
肌無力的分佈、發作時的年齡、家族史、足畸形(高足弓和杵狀趾)和神經傳遞速度的檢查結果可幫助醫生鑒別夏科-馬里-圖思病和德-索氏病以及其他原因所致的神經病。沒有治療能阻止這些疾病惡化。戴足托能幫助矯正足下垂,有時,需要作矯形手術。
-脊髓性肌萎縮
脊髓性肌萎縮是脊髓和腦幹的神經細胞變性引起進行性肌無力和萎縮的遺傳性疾病。
癥狀
癥狀首先出現在嬰兒期和兒童期。急性脊髓性肌萎縮(韋德尼希-霍夫曼病)所致的肌無力出現在2~4個月的嬰兒中,這種病是常染色體隱性遺傳病,也就是說需要從父母雙方分別獲得一個隱性基因。
患有中度脊髓性肌萎縮的小孩在一年或兩年內保持正常,然後出現肌無力,下肢比上肢更重。此病通常不累及呼吸、心臟或顱神經,病程也緩慢。
慢性脊髓性肌萎縮(沃-庫-韋氏病)開始於2~17歲的青少年,病情緩慢加重,因此,此病患者比患其他類型的脊髓性肌萎縮者壽命更長。肌無力和萎縮開始於雙下肢,隨後擴展到雙上肢。
診斷和治療
當兒童出現不能解釋的乏力和肌萎縮時,醫生應考慮到這些少見的疾病,因為這些病都是遺傳性的,家族史可以幫助作出診斷。
這類疾病沒有特異性的治療方法。物理治療、戴足托和其他特殊裝置有時可能有幫助。
基本信息
- 中文名
- 周圍神經疾病
- 就診科室
- 五官科
- 含義
- 神經損害的總稱
- 分類
- 神經痛和神經病
- 結果
- 功能障礙