共找到2條詞條名為佩梅病的結果 展開
- 佩梅病
- 佩利措伊斯-梅茨巴赫病
佩梅病
佩梅病
佩梅病(Pelizaeus-Merzbacher disease,PMD)是一種罕見的瀰漫性腦白質髓鞘形成障礙的X 連鎖隱性遺傳疾病,屬蛋白脂蛋白1(proteolipid protein l,PLP1)相關的遺傳性髓鞘形成障礙疾病譜中的一種。PMD 特徵性病理改變為神經髓鞘不能正常形成,而非其他遺傳性白質腦病那樣脫髓鞘改變。
1885 年,Pelizaeus 率先報道了有5 例男性患兒的家系,主要表現為眼球震顫、四肢麻痹、共濟失調、發育遲緩等。Merzbacher 於1910 年再次對Pelizaeus 所報道的家系進行研究,此時受累的患者有14 例,有2 例女性患者,結果發現,此病具有X 連鎖隱性遺傳特徵且在腦組織活檢中發現白質髓鞘缺失故,將此病命名為PMD。此病病理表現為髓鞘區與脫髓鞘區交錯,呈虎斑樣外觀,鏡下可見嗜蘇丹樣物質沉積於半卵圓中心、腦幹和小腦內。PMD是嚴重的致死、致殘性神經遺傳病,患者壽命均較短,嚴重者僅能存活至幾歲,甚至生后即死亡。其發病率在美國為1/500 000~1/300 000, 我國尚缺乏相關的發病率研究,2007年本課題組首次在國內對一個PMD大家系進行了PLP1基因診斷與報道,並於2006年至今臨床診斷PMD 76例,完成PLP1基因突變分析60例(其餘患者基因分析正在進行之中),其中基因確診57例。以下將從PMD病因、臨床表現、輔助檢查、診斷、、鑒別診斷、治療、產前診斷與遺傳諮詢進行闡述。
PMD 的致病基因為位於Xq22.2 的蛋白脂蛋白1(proteolipid protein 1,PLP1)基因,PLP1基因全長約17 kb,含有7 個外顯子,編碼含有276 個氨基酸的PLP1 蛋白和其剪切異構體DM20。PLP1 是中樞神經系統髓鞘的主要成分,約佔整個髓鞘蛋白的50%。其主要功能是組成並穩定髓鞘同時對少突膠質細胞前體細胞的發育起重要作用。PLP1主要在少突膠質細胞(oligodendrocyte, OL)表達,少突膠質細胞是神經膠質細胞的主要類型,遍佈於中樞神經系統的灰質與白質內,尤以白質為多,少突膠質細胞是髓鞘形成細胞,少突膠質細胞正常發育為中樞神經系統髓鞘的完整性提供了保障。髓鞘是包裹於神經細胞軸突外面的管狀外膜,髓鞘上有朗飛氏結,可使神經衝動跳躍傳遞,髓鞘由髓磷脂構成具有絕緣作用,能夠提高神經衝動的傳導速度,並具有軸突保護作用,軸突傳導速度由突觸直徑、髓鞘厚度、朗飛氏結的數目與空間分佈及結間區域離子通道的分子組成進行調控,髓鞘在神經信息精確、高效傳遞以及中樞信息整合的發揮中起著極其重要的作用。少突膠質細胞/髓鞘異常能夠改變軸突束的穩定,從而影響神經細胞的基本電傳導模式,最終影響正常突觸傳遞。研究表明Plp過表達轉基因小鼠可以造成認知行為損害,可能與少突膠質細胞/髓鞘功能障礙通過改變谷氨酸能與多巴胺能信號傳導導致的神經環路異常有關,在少突膠質細胞/髓鞘異常的神經元中進行電生理的相關研究將有助於理解這些信號通路中分子在軸突-髓鞘的相互作用機制。
PLP1 基因缺陷可使PLP1 蛋白表達過度(PLP1 基因重複突變)、表達下降或細胞內分佈異常(PLP1 基因點突變)以及PLP1缺失,這些均可以導致少突膠質細胞/髓鞘功能異常,從而導致髓鞘形成異常和(或)少突膠質細胞死亡,使得廣泛白質區域髓鞘缺乏或減少。PLP1基因不同突變類型通過不同的細胞與分子機制導致了臨床表型的差異。
PMD 的典型臨床表現為眼球震顫、肌張力低下、共濟失調及進行性運動功能障礙。在疾病發展過程中,多數患兒初始能逐漸進步,然後出現智力運動發育逐漸倒退,且運動功能障礙比智力障礙更顯著。PMD屬於PLP1 相關性疾病中的一種,PLP1 相關性疾病是一個由重到輕的連續性疾病譜,按臨床表現從重到輕和起病年齡的不同分為6 型(表1):先天型PMD(connatal PMD),經典型PMD(classic PMD),中間型(a transitional form),無PLP1 綜合征(PLP1 null syndrome),複雜型痙攣性截癱(complicated spastic paraplegia)和單純型痙攣性截癱(uncomplicated spastic paraplegia)。
先天型PMD出生時起病,臨床癥狀重。表現為鐘擺狀眼震、肌張力低下、吞咽困難、喘鳴,部分患兒可有癲癇發作。認知功能嚴重受損,語言表達嚴重受累,但可有非語言交流,部分患兒有理解語言的可能。整個發育過程中不能獨自行走。隨病程進展肢體逐漸痙攣。多數於兒童期死亡,少數存活時間較長,但一般不超過30 歲。
經典型PMD為Pelizaeus 和Merzbacher所描述的PMD,也是最常見的一種。多於生后數月內發病,最遲不超過5 歲。早期表現有眼球震顫、肌張力低下。10 歲前運動功能可緩慢進步,可獲得上肢隨意運動和行走能力,之後逐漸倒退,隨病程進展眼球震顫可消失,繼而出現運動發育障礙,如步態蹣跚、共濟失調、四肢癱瘓等,還可伴認知功能損害和錐體外系異常表現。患者多在30~70 歲死亡。
中間型的臨床表現介於先天型和經典型之間。
此種類型比較特殊,沒有眼球震顫表現。1 歲內發育多正常,於1~5 歲起病。主要表現為輕度四肢痙攣性癱瘓,共濟失調,輕至中度認知功能受損,可獲得一定語言功能,多於青春期后出現倒退,部分患兒可伴有輕微周圍神經癥狀。壽命多在50~70 歲。
患兒1 歲內多發育正常,多於1~5 歲起病。主要表現為眼球震顫,共濟失調,下肢進行性無力和痙攣,自主功能紊亂(如膀胱痙攣),無或輕微認知功能受損,語言功能多存在。壽命多在40~70 歲。
本型為最輕的一種類型,患兒1 歲內通常發育正常,1~5 歲起病,也可以30~40 歲才出現癥狀。主要表現為逐漸出現的下肢進行性無力和痙攣,自主功能紊亂(如膀胱痙攣)。但患者無眼球震顫和認知功能受損。壽命多正常。
表1 PLP1相關性疾病臨床分型
| 表型 | 發病年齡 | 神經系統表現 | 行走 | 語言 | 死亡年齡 |
| 先天性PMD | 新生兒期 | 生后即發現眼球震顫、吞咽無力、喘鳴、肌張力低下、嚴重痙攣伴/不伴驚厥、認知障礙 | 從不會 | 不會、也可能有非語言交流與語言理解 | 嬰兒到30歲 |
| 經典性PMD | 5歲以內 | 生后兩月內發現眼球震顫、起始肌張力低下、痙攣性截癱、共濟失調、步態不穩伴/不伴肌張力障礙、手足徐動症、認知障礙 | 輔助行走,兒童/青春期喪失行走功能 | 通常可以出現 | 30~70歲 |
| 無PLP1綜合征 | 5歲以內 | 無眼球震顫、輕度痙攣性截癱、共濟失調、周圍神經病、輕到中度認知障礙 | 會 | 會,通常青春期惡化 | 50~70歲 |
| 複雜SPG2 | 5歲以內 | 眼球震顫、共濟失調、自主神經功能障礙*、痙攣性步態、輕度或無認知障礙 | 會 | 會 | 40~70歲 |
| 單純SPG2 | 通常為5歲以內,也可以30~40歲 | 自主神經功能障礙*、痙攣性步態、無認知障礙 | 會 | 會 | 正常 |
註:中間型PMD臨床表現介於先天性與經典型PMD之間;*痙攣性膀胱
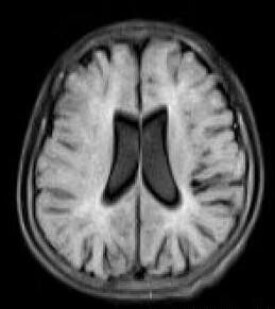
magnetic resonance imaging,MRI,PMD 影像學主要表現是髓鞘發育不良或髓鞘完全無發育。頭顱MRI可顯示髓鞘化異常,主要表現在腦白質T2 加權像和Flair 像瀰漫性高信號,此項檢查對PMD 診斷具有重要意義。因為生后第1、2 年是腦白質髓鞘化形成的重要時期,所以此時頭顱MRI 表現特異性相對較小。但因正常3 個月嬰兒的內囊後肢、胼胝體壓部和視放射區已經有髓鞘形成,所以早期這些部位異常對PMD 診斷具有重要意義。隨著PMD 患兒年齡逐漸增加,其腦白質發育極其落後,頭顱MRI 表現為新生兒樣腦白質表現,T1 加權像腦白質改變常不明顯,T2 加權像腦白質幾乎全部為高信號。隨著病情進展,腦白質容積縮小,表現為胼胝體變薄,腦室擴大和皮質內陷。痙攣性截癱患兒腦白質異常程度較PMD輕,其頭顱MRI T2 加權像可表現為片狀高信號。
magnetic resonance spectrum,MRS,膽鹼複合物(choline,Cho)包括甘油磷酸膽鹼、磷脂醯膽鹼、磷酸膽鹼等,是細胞膜磷脂代謝的重要組成成分。當細胞膜分解破壞時,Cho 水平會升高。因為PMD 是髓鞘形成障礙性疾病,Cho 水平不高,與腦白質脫髓鞘疾病比較具有重要意義。Bonavita 等報道無PLP1 綜合征中N-乙醯天門冬氨酸(N-acetylaspartate,NAA)水平下降。相反,在有PLP1 基因重複突變患兒中NAA 水平會增高,此項容易和Canavan 病相混淆。
PLP1基因突變類型多樣。截至目前已發現PLP1相關疾病的PLP1 基因突變142 種(human gene mutation databases),包括重複突變、點突變與缺失突變。以重複突變最為常見,佔PMD 患者總數的50%~70%,點突變佔PMD患者總數的10%~25%。據此,在臨床診斷為佩梅病患者基因檢測策略首先進行PLP1基因重複突變檢測,多重連接探針擴增技術(multiplex ligation-dependent probe amplification ,MLPA),是近幾年發展起來的一種針對待檢DNA序列進行定性和半定量分析的新技術。該技術高效、特異,在一次反應中可以檢測30-48個核苷酸序列拷貝數的改變,目前已經應用於多個領域、多種疾病的研究。在佩梅病診斷中用於檢測PLP1基因重複/缺失突變。結果正常者應用DNA直接測序方法進行點突變的檢測。
對於臨床疑似PMD患兒需行PLP1 基因檢查以確診。
研究表明在PLP1相關疾病譜系中基因型與表型具有明顯的相關性: PLP1 基因突變以重複突變最為常見,佔50%~70%,點突變佔10%~25%,而缺失突變僅佔2%左右。 PLP1基因重複突變見於大多數經典型與中間型PMD;點突變臨床表型分佈廣泛,可以見於PLP1相關疾病的全部臨床表型,但以先天型PMD為多;而缺失突變見於無PLP1綜合征與痙攣性截癱2型。我們課題組對53例完成PLP1基因突變分析PMD患者的實驗結果顯示:71.7%為PLP1基因重複突變,其中臨床表型為經典型與中間型PMD分別佔68.4%(26/38)與26.3%(12/38);22.6%為點突變,其中41.6%(5/12)為先天型PMD;5.7%未發現PLP1基因改變。最新研究還表明PMD患者伴隨PLP1基因重複突變的X染色體所累及區域拷貝數變異(copy number variation, CNV)片段大小與臨床表型密切相關,我們前期應用基因晶元對上述38例PLP1基因重複突變患者的相關區域CNV的改變進行初步分析結果也顯示了CNV片斷的大小和模式與臨床表型密切相關。迄今為止北京大學第一醫院兒科通過基因檢測共確診58 例PMD 患兒。
PMD 臨床診斷主要依據典型臨床表現和頭顱影像學檢查,確診依靠分子遺傳學研究。臨床上遇到男性患兒,以眼球震顫起病,主要表現為眼球震顫,肌張力低下、共濟失調及進行性運動功能障礙,頭顱MRI 示T2 加權像腦白質瀰漫性高信號,要考慮PMD的可能,應進一步行PLP1 基因檢查以確診。
如果PLP1 基因檢查無異常,應該進一步查GJA12基因,尤其對於臨床表現為經典型PMD 的女性患兒。
佩梅樣病(Pelizaeus-Merzbacher-like disease,PMLD)是一種少見的常染色體隱性遺傳的瀰漫性腦白質髓鞘形成障礙疾病,其臨床表現和PMD 患者相似,故得名PMLD。目前已知的PMLD 的致病基因是縫隙連接蛋白α12(gapjunction protein alpha 12,GJA12),又稱GJC2,還有其它基因可以引起PMLD的臨床表現,因此,將GJA12/GJC2導致的PMLD叫做PMLD1,此基因是2004 年由Uhlenberg 等確定。GJA12 基因長約9.9 kb,包括2 個外顯子,編碼區位於第2 外顯子,基因編碼產物為縫隙連接蛋白47(gap junction protein 47,connexin 46.6,Cx47),GJA12 基因突變可導致嚴重神經系統病變。PMLD 發病機制尚不清楚,目前認為PMLD 相關的GJA12 基因突變,可能導致Cx47表達變化,干擾了星形膠質細胞(astrocytes)與少突膠質細胞(oligodendrocytes)之間的偶聯。星形膠質細胞和少突膠質細胞之間通過縫隙連接相互偶聯。不同細胞表達不同的縫隙連接蛋白。星形膠質細胞之間通過Cx43/Cx43 和Cx30/Cx30 通道相偶聯。星形膠質細胞/少突膠質細胞之間通過Cx43/Cx47 和Cx30/Cx32 通道相偶聯。免疫染色切片顯示Cx47 在少突膠質細胞表達,且靠近其細胞邊緣,GJA12基因的錯義突變導致了Cx47 功能的缺失,因此認為GJA12基因突變影響Cx43/Cx47 介導的A/O 偶聯,從而出現一系列臨床表現。北京大學第一醫院兒科2007 年在國內率先診斷並報道了2 例PMLD,基因分析結果示1 例為GJA12 基因點突變,1 例為移碼突變,且為1 號染色體父源性單親二倍體所致。截至到目前為止,本課題組臨床診斷9例PMLD患者,基因確診的PMLD1患者4例,國際上基因確診僅60餘例。
PMLD 與PMD 臨床表現相似,頭顱MRI表現 與PMD 基本一樣,難以區分,但PMLD 患者出現驚厥機率大。PMLD 是常染色體隱性遺傳,男女發病無顯著差別為常染色體隱性遺傳,男女發病沒有明顯差別,但PMD 是X 連鎖隱性遺傳,男性多見,且更嚴重。根據一般的影像學及生化檢查很難將此兩種疾病分開,目前只能依賴基因突變分析進行確診,如果PLP1 基因檢查無異常,應該進一步查GJA12/GJC2基因檢測,尤其對於臨床表現為經典型PMD 的女性患兒。
vanishing white matter disease,VWM,此病也可表現為大腦白質的瀰漫性受累,但是異常白質可以出現液化表現,在Flair 像能看得很清楚似腦脊液信號。臨床表現上也無眼震。而是以運動障礙起病、運動障礙重於智力障礙,每遇感染所致發熱或輕微的頭部外傷可引起病情明顯加重,可以有共濟失調、癲癇發作和視神經萎縮。
是由溶酶體中N-乙酸神經氨酸(NANA)蓄積所致的遊離唾液酸貯積病,臨床上也可表現為肌張力低下、眼球震顫和智力運動發育遲緩。但是癲癇在此病中較PMD 中更常見,而且此症可有粗陋面容、肝脾大以及心臟擴大。頭顱MRI 在較重的患兒中可表現為瀰漫性T2 高信號,在相對輕的患兒中主要集中在腦室周圍。高效液相串聯質譜分析檢測尿中遊離唾液酸增加可以診斷,培養的皮膚成纖維細胞中遊離唾液酸貯積在溶酶體而不是胞漿中或者發現SLC17A5 基因的致病突變可確診此病。
目前,佩梅病及佩梅樣病尚無滿意治療方法。妊娠婦女如果懷疑是PLP1 或者GJA12 基因突變攜帶者,可進行遺傳諮詢、產前診斷。但受累胎兒的表現型很難準確預測,因為帶有同樣突變的家族成員其表現型可以差別很大。隨著幹細胞研究的深入,有些疾病已經實現了用幹細胞移植來治療。雖然目前仍有一些關鍵的技術問題還沒有解決,但是相信在不久的將來用幹細胞移植治療PMD 及PMLD也會成為可能。
產前診斷(prenatal diagnosis)又稱宮內診斷或出生前診斷,它是在遺傳諮詢的基礎上,利用各種診斷技術,對胎兒疾病做出宮內診斷。產前診斷除了對遺傳性疾病進行產前診斷,主要通過遺傳學檢測和影像學檢查,對高風險胎兒進行明確診斷,通過對患胎的選擇性流產達到胎兒選擇的目的,從而降低出生缺陷率,提高優生質量和人口素質。
產前診斷方法依據取材和檢查手段的不同,一般分為侵入性(invasive)和非侵入性(noninvasive)兩大類, 前者主要包括羊膜腔穿刺、絨毛取樣、臍血取樣、胎兒鏡和胚胎活檢等;後者包括超聲波檢查、母體外周血清標誌物測定和胎兒細胞檢測等。目前產前診斷中仍以侵入性方法為主,以羊膜腔穿刺和絨毛取樣兩種最常用。在佩梅病家庭中先證者診斷明確后,進行家系分離分析,明確該家系中的遺傳方式,即可進行遺傳諮詢與產前診斷,先證者經分子檢測確定診斷,先證者之母再次懷孕后,簽署知情同意書,採集胎兒絨毛或者羊水提取DNA進行產前分子診斷,SRY用於胎兒性別鑒定,DXS6797、DX6807與AR分別為X染色體上短重複序列標籤進行是否有母血污染與胎兒生物學父母的檢測,對於基因診斷則依據先證者分子遺傳學診斷所用方法來進行。
遺傳諮詢(genetic counseling)是一個幫助人們理解和適應遺傳因素對疾病的作用及其對醫學、心理和家庭影響的程序。這一程序包括:通過對家族史的解釋來評估疾病的發生或再發風險率;進行相關疾病的遺傳、實驗室檢測、治療處理及預防的教育,並提供與疾病有關的各種可以求助的渠道和研究方向;輔導促進知情選擇和對所患疾病及其再發風險的逐步認知和接受。對於具有產前診斷指征者均需進行遺傳諮詢。
基本信息
- 中文名
- 佩梅病
- 外文名
- Pelizaeus-Merzbacher disease,PMD
- 季節分佈
- 四季
- 是否傳染病
- 否
- 癥狀表現
- 鐘擺狀眼震、肌張力低下、吞咽困難、喘鳴
- 就診科室
- 內科
- 傳染性
- 無傳染性