不自主運動
不自主運動
不自主運動或稱異常運動,為隨意肌的某一部分、一塊肌肉或某些肌群出現不自主收縮。是指患者意識清楚而不能自行控制的骨骼肌動作。臨床上常見的有肌束顫動、肌纖維顫搐、痙攣、抽搐、肌陣攣、震顫、舞蹈樣動作、手足徐動和扭轉痙攣等。
不隨意運動(involuntarymovement)是患者意識清醒時(有時也可發生在輕度意識障礙或淺昏迷時)出現不能控制的骨骼肌不正常運動,表現形式多樣,一般在情緒激動時加重,睡眠時停止;其病變大多發生在錐體外系大腦皮質運動區腦幹、小腦脊髓周圍神經甚至肌肉病變時也可引起。錐體外系涉及所有錐體系以外與運動調節有關的結構及下行通路,包括基底核小腦及腦幹中諸多核團錐體外系癥狀通常指基底核病變導致的姿勢、運動異常。與基底核病變有關的不隨意運動的解剖及生理學基礎簡介如下: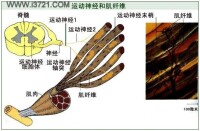
不隨意運動
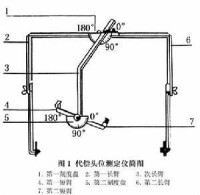
基底核中與運動功能有關的主要結構是紋狀體其組成及病變綜合征如圖1。
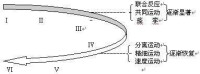
紋狀體與大腦皮質及其他腦區的纖維聯繫相當複雜(圖2),與運動皮質的聯繫環路是基底核實現運動調節功能的主要結構,包括:
1.皮質-新紋狀體-蒼白球(內)-丘腦-皮質迴路。
2.皮質-新紋狀體-蒼白球(外)-丘腦底核-蒼白球(內)-丘腦-皮質迴路。
大腦皮質運動區及其下行纖維、基底節、腦幹、小腦、脊髓、周圍神經以及肌肉各部的病變均可引起不自主運動。如舞蹈樣動作、手足徐動、扭動痙攣由新紋狀體病損引起;節律性與局限性肌陣攣與下橄欖核、齒狀核及紅橄欖束的損害有關:舞動運動為對側腦底核的病變所致;震顫可由紋狀體、小腦及小腦有關的結構或額葉病變引起;肌束顫動見於運動神經元病變等。不自主運動可因生理或精神因素引起,但大多為器質性病變所致,主要見於感染、中毒、變性、遺傳和家族性發育異常等疾患,也可見於腦血管病、外傷、腫瘤等。
近年對神經系統遞質(化學傳遞物質)的研究有很大進展認為該物質貯存於神經軸突末梢的突觸囊泡中其中包括乙醯膽鹼、去甲腎上腺素多巴胺、5-羥色胺、γ-氨基丁酸和甘氨酸等如多巴胺以抑製為主乙醯膽鹼則以易化為主。
錐體外神經元的功能狀態取決於遞質的平衡,多巴胺與乙醯膽鹼則更為重要如紋狀體的多巴胺下降與乙醯膽鹼失去平衡則出現震顫麻痹,5-羥色胺與組胺也與震顫的產生有關。又如多巴胺的易化作用增強則會出現不自主的舞蹈動作目前對這一方面的機理尚未完全闡明錐體外系病變導致的姿勢、運動異常,出現不隨意運動的類型主要包括:
1.帕金森病患者可出現靜止性震顫(statictremor)。
2.帕金森病時伸肌與屈肌張力均增高可導致肌強直(rigidity)。
4.Huntington舞蹈病Wilson病、肝性腦病Hallervorden-Spatz病、酚噻嗪類及氟哌啶醇慢性中毒等可導致手足徐動症(athetosis);偏側手足徐動症多見於腦卒中。
5.偏身投擲運動(hemiballismus)為丘腦底核及與其聯繫的蒼白球外側部急性病變所致。
6.錐體外系病變(如肝豆狀核變性)多可出現肌張力障礙(myodystonia)。
7.抽動-穢語綜合征多數學者傾向認為是一種特殊的癲癇類型。
臨床表現多種多樣現就其類型及特點分述如下:1.震顫是身體某一部分循一定方向的節律性來往擺動且幅度大小不一的不隨意運動,於手部最常見,其次為眼瞼頭部和舌。震顫又可分為生理性、功能性和病理
不隨意運動
(2)功能性震顫:此型震顫可能是生理性震顫的加強。這是一種細小快速而無規律的震動振幅大小不等震顫常不規則與多變。多見於手指無肌張力的改變往往與精神因素有關。甲狀腺功能亢進焦慮疲勞所引起的手部震顫也屬此類這是由於腎上腺素作用於肌梭提高其敏感性以及交感神經活動增強的結果。
(3)病理性震顫可分為以下幾種:
(一)面肌抽搐(facialtic,Beu'sspasm)由於疼痛刺激引起面部肌肉反射性痙攣性收縮者稱“痛性抽搐”,見於三叉神經痛。當疼痛發作時常伴有患側面肌反覆發作性抽搐,口角牽向患側,結膜充血,流淚等癥狀。此外尚有一般的面肌抽搐,多為一側性,表現為眼瞼抽搐、伴有皺眉肌、鼻部諸肌、頰肌收縮,眼瞼呈快速頻繁的抽動,如病程過長,反覆發生可引起眼瞼強直性收縮,提上瞼肌收縮,瞼裂縮小。面肌抽搐和面肌痙攣臨床表現相同。
(二)手足抽搦症(tetang)由低血鈣或中毒引起。多見於嬰兒、兒童與哺乳期婦女。表現為間隙發生的雙側強直性痙攣,以上肢顯著。典型的“助產手”包括手指伸直並齊,掌指關節屈曲,大拇指對掌內收,腕部屈曲,常伴有肘部伸直和外旋。牽涉下肢時,有足趾和踝部的跖曲和膝部伸直。嚴重時可有口、眼輪匝肌痙攣。發作時意識清晰。Chvostck征和束臂試驗陽性。低血鈣可能同時產生手足抽搦和癇性抽搐。
(三)破傷風(tetanus)破傷風患者受到外界刺激時,常發生全身強直性或陣攣性的抽搐,歷時數秒鐘。意識清醒,在抽搐間歇肌肉並不鬆弛,捫診仍可發現堅硬的強直狀態,以咀嚼肌最為顯著。外傷病史、痙笑病容,角弓反張等均可提示破傷風的診斷。
(四)新生兒抽搐(neonatalconvulsion)新生兒抽搐均屬癇性抽搐。輕者呈局限性或偏側性陣攣性痙攣,腦電圖顯示一側尖波灶或正常。重症者可呈全身強直性痙攣或肌陣攣,或是微小的眼球或肢體強直性或陣攣性動作,伴有腦電圖上的多發性尖波灶,瀰漫性陣發性活動或低平電位。
(五)癲癇大發作(grandmalepilpsy)臨床表現為突然意識喪失,頭轉向一側或後仰,眼球向上或一側移動,上肢伸直或屈伸,手緊握,下肢伸直或表時屈曲。由於呼吸肌強直收縮,可出現呼吸暫停,面及皮膚緊紺,瞳孔散大,對光反應消失。繼而全身肌肉由持續的收縮轉為一張一弛的陣攣性收縮,頭部、軀幹及四肢均呈強烈的收縮。由於腹壁肌及膀胱收縮,可有小便失禁,發作停止后即轉入昏睡狀態。醒后除先兆外,對發作經過不能回憶。自發作開始到意識恢復經歷5~15分鐘。發作間隙期腦電圖可有各種癇樣發放。
(六)癲癇局限性發作(localiedepilepsy)為一系列的局部
不隨意運動
(八)癔前些天性抽搐(hystenicalconvulsion)發作前有精神因素,多於他人在場時發病,發作時突然倒下,頭部後仰,全身僵直,牙關緊閉,握拳時大拇指在掌外。強直性痙攣后,繼有不規則的手足舞動。常雜以捶胸頓足,哭笑叫罵等情感反應。無意識喪失,瞳孔和跖反射等無變改。而色如常或潮紅,大多無咬舌、跌傷和大小便失禁,每次發作歷時數十分鐘到數小時,經他人撫慰或治療而止。患者能描述發作經過。
(一)書寫痙攣(writingspasm)此病多屬於神經功能性疾病。是在書寫時由於手肌緊張及運動異常而導致書寫功能障礙。患者在持筆時或開始寫字時困難。這種痙攣現象主要發生在手指、腕部甚至整個上肢。有的病例引起固有伸肌呈痙攣性強直收縮,繼之沿食指及中指漸漸蔓延到固有伸拇指長肌收縮,產生拇指外展。此病最重要的特點是書定時痙攣,不寫字時癥狀消失。
不隨意運動
1.神經性肌強直(neuromyotnia):病因不明,從5歲到60歲之間皆有發病。病情常在數月或數年內逐漸進展並加重。在靜止狀態、活動狀態時肌肉均呈持續性收縮,伴有劇烈疼痛。常首先侵犯手足,以後向面部及咽喉部肌肉、舌肌、肢體、軀幹肌肉蔓延,伴有廣泛的肌肉顫搐、常發生窒息,並由於吸入性肺炎而死亡。
2.MCArdle氏徵候群(MCArdlessymdrome):由於肌肉缺乏磷酸化酶或磷酸果糖激酶而使肌肉收縮不正常,呈痙攣性收縮,由於強直痙攣收縮,被動牽引而產生疼痛,疼痛隨運動度增加而加重,客觀檢查無異常發現。
3.僵人綜合征(stiffmansyndrome):常見於男性,有軸性肌肉針刺樣感的疼痛,然後發生對稱性的持續的骨骼肌僵硬,抽搐,痛性肌痙攣。眼肌、心肌與平滑肌不受影響,肌僵硬不受睡眠、脊髓麻醉和巴比妥類藥物的影響。
(一)靜止性震顫(statictremor)在肌肉完全鬆弛的情況下,即安靜狀態下出現的震顫叫靜止性震顫。這種震顫於睡眠時消失,運動時減輕或消失。震顫節律為4~6次/秒,比較有節律。靜止性震顫見於中腦被蓋近結合臂交叉部位病變。
1.震顫麻痹(paralysisagitans):或稱帕金森氏綜合征(parkinson'ssyndrome)震顫麻痹患者的靜止性震顫常幅度較小,有節律(4~6次/秒)。一般情況下震顫發后的順序是先在一側,其次是同側下肢,再次是另一側上肢而波及到下肢。下頜、口唇、舌和頭部均最後受累。上肢震顫常比下肢重。手指的節律性震顫形成所謂:“搓丸樣動作”、在本病早期,震顫僅於肢體的靜止狀態下出現,作隨意運動時減輕或消失,情緒激動時加重,睡眠時完全停止。伴有肌強直和運動減少。
靜止性震顫是本病的特徵之一,但當此病的進展期,其無論靜止時或運動時均出現震顫,則震顫往往在上午重,清晨輕,睡眠時消失。並有既往病史和相應的狀態,如腦炎后Parkinsson綜合征過去有腦炎病史常見有動眼危象,皮脂溢出及流涎增多。
2.老年性震顫(seniletermor):發生於老年人,震顫常見於靜止時。其頻率、幅度與Parkinson綜合征類同。震顫以頭、下頜與唇部多見,呈點頭狀或左右側向震顫。但無肌張力增強及運動減少。
3.肝豆狀核變性(hepatolenticulardegeneration):又稱Wilson病。本病的震顫大致與parkinson綜合征相類似,主動肌與拮抗肌交互性節律收縮,當隨意運動
不隨意運動
1.小腦疾患:小腦病變時主要為意向震顫,系齒狀核或發自齒狀核通路病變所致。靜止時不出現震顫,運動時震顫速度不快,但振幅粗大,不規則。如作鼻試驗或舉杯時,震顫大為明顯。伴有運動障礙。
2.多發性硬化(multiplesclerosis):本病以小腦性意向性震顫為多,常為意向性震顫和姿勢性震顫合併出現。患者在靜止時不出現震顫,而當坐位或立位時頭現軀幹前後搖動,振幅開始小,以後逐漸增大,結合反覆發作的腦、脊髓和視神經受損的特徵可考慮診斷。
(三)姿勢性震顫(posturaltrema)為身體受累部分主動的保持某種姿時出現的震顫。即在靜止狀態下不出現,只有當患者身體處於某種姿勢的情況下才出現震顫。臨床上常見疾病有:1.特發性震顫(idiopathictrema):起病早且常有震顫家族史。姿勢性震顫為本病的唯一癥狀。往往見於手部,影響精細動作、書寫和用匙筷進食。其次為頭部,表現為細小的點頭或搖頭動作。患者平舉上肢時,可見雙側指部和腕部的5~8次/秒震顫,振幅不定,在精神緊張時加劇。本病既無肌強直及運動減少,又無肌張力低下、共濟失調、眼震和醉漢步態等癥狀易與震顫麻痹和小腦疾患相鑒別。
2.撲翼震顫:主要特點產姿勢性震顫。在靜止與睡眠時消失,精神興奮時增強。當患者兩上肢前伸,和腕部伸直維持一定姿位時,腕關節突然屈曲,而後腕關節可迅速伸直至原來的位置,這樣反覆的腕關節屈曲及伸直,狀如撲翼。這種震顫以不規則、無節律、振幅經常有變化為特徵。主要見於全身性代謝疾病,如肝昏迷、尿毒症、慢性肺功能不全、低血鉀、心力衰竭時的紅細胞增多症、脂肪痢、Whipple病及低血鎂症。系中樞神經系統反覆抑制和興奮肌張力所致。
3.中毒和感染性震顫:各種藥物和酒精、鋰、鉛、磷、鎂、汞、一氧化碳等中毒以及各種感染(如傷寒、水痘、天花)均可發生姿勢性震顫。如酒精中毒時的這種震顫於靜止時不明顯,當上肢前伸手指分開並保持此姿勢時震顫明顯,以震動性震顫為其特徵。如發生於手指,進而波及舌、面肌和鼻翼肌。在笑和說話時可以見到細小的震顫。汞中毒引起的震顫,呈中等節律震顫,靜止時出現,運動時更突出,越近目的地進震顫越強。即靜止性震顫與姿勢性震顫混合出現,當疲勞與精神興奮時增強。
(四)混合性震顫同時具有上述兩種或兩種以上的震顫稱混合性震顫,如小腦病變、肝豆狀核變性可合併發生靜止性、姿勢性及意向性震顫。癔症性震顫表現為多變不規則的震顫,震顫大小不一。靜止和運動時都存在。有明顯的精神因素,常伴有其他癔症性癥狀,分散病人注意時震顫緩解,集中注意於病變部位時震顫增強。
(一)節律性肌陣攣
1.軟齶、喉部肌及其它部位節律性肌陣攣:在軟齶、咽、喉部肌群以及眼肌、膈肌等產生肌收縮,呈同步性每秒2~3次,每分鐘可快達100次左右。收縮可呈持續性,睡眠或鎮靜劑不能控制,針刺或興奮劑可改善癥狀,隨意運動時可暫時停止。最常見的一型是軟齶震顫局限於軟齶部,軟齶作上下有節律的顫動,伴有回及喉的運動,肌陣攣可擴延至眼肌、面肌及膈肌。如膈肌陣攣時出現呃逆,眼外肌陣攣時出現眼球偏視性節律運動。此種肌陣攣是由於延髓橄欖、中腦紅核與同側小腦齒狀核三個部位聯結的三角區病損的結果。常見於血管性病變、腫瘤和腦炎等。
2.流行性腦炎時的節律性肌陣攣:在流行性腦炎的急性期或後遺症期,出現的一處肌陣攣。這種肌陣攣呈快速的節律,約每分鐘10次左右的收縮,呈持續性,

不隨意運動
(1)遺傳性肌陣攣(Unverricht氏病):有家族史,主要表現為突然,閃電樣肌肉收縮,一般為雙側,亦可見於單側,常侵犯頸、肩、臂肌。發作次數隨年齡增長而減少,發作前有前驅癥狀,如心悸、心前區不適感等。肌陣攣常因情緒因素、聲光刺激、疲勞、寒冷加重。可伴有癲癇大小發作等。
(2)持續性局限性肌陣攣:一塊肌肉或一群肌肉引起陣攣性收縮,產生的部位以肢體遠端為多。多數情況下呈節律性抽動,常與Lachson氏癲癇相混淆,所不同者前者呈持續性。
(3)小腦性肌陣攣性癲癇:本病有癲癇、肌陣攣、小腦癥狀三大主征。不產生運動效果。此型肌陣攣當隨意運動時出現反抗運動,即由於拮抗肌的作用產生相反方向的運動。可發生於兒童隱性神經母細胞瘤和成人支氣管肺癌的患者。
(二)非節律性肌陣攣
1.多發性副肌陣攣:1881年Friedrich氏首先報道故又名Frid—erich氏病。肌肉出現陣攣性收縮,不同步,無節律,一般運動效果不明顯,收縮頻率為每分鐘40~50次。在情緒影響下出現劇烈收縮,多呈進行性,安靜時消失。主要侵犯肌肉和軀幹肌。常先在下肢出現肌陣攣,以後波及身體其它部位,面肌和呼吸不常受累。腱反射、感覺無異常,不出現癲癇和肌萎縮。2.肌肉纖維性收縮(Morvan氏纖維性舞蹈病):患者開始先在腰部及下肢有疲勞感和肌肉疼痛。肌纖維性收縮多在大腿後部,以後可波及軀幹及上肢,不侵犯頭面部,以肢體近側端明顯。肌肉纖維性收縮是在肌肉的一部分呈現小的波動運動,與肌纖維震顫不同點在出現部位和頻度不同,本病的好發部位主要是下肢,而軀幹和上肢少見。在靜止時、睡眠時收縮不消失,機械刺激不能誘發。目前認為本病與間腦病變有關。
(一)小舞蹈病(xhoreaminor)主要病理改變為基底節、大腦皮質、腦幹、小腦等處散在的動脈炎和神經細胞變性。常為急性風濕病的一種表現。80%發病於兒童期,多呈急性起病,起飛現前常有前驅期。此期平均一周左右,有輕度體溫增高。以後出現四肢及脊背疼痛;手足活動笨拙不靈。持物不穩定,繼之出現舞蹈樣運動。開始輕而局限,逐漸波及全身,其舞蹈樣運動上肢表現為各關節交替發生伸直、屈曲、扭轉等動作。手指不停的屈曲與內收;伸手和舉臂過頭時出現特殊姿勢,腕關節屈曲,掌指關節過伸,各指伸直分開,手臂過度旋前,稱旋前肌征(pronatorsign)。下肌表現為步態顛簸,常常跌倒。軀幹亦可繞脊柱捲曲或扭轉。面部表現為扮鬼臉、皺額、眨眼、撅嘴、吐舌、擠眉等等,變幻不已。呼吸可因軀幹和腹肌的不自主運動而變為不規則。舞蹈樣運動可因情緒激動或作自主運動而加劇,平卧安靜時減輕,睡眠時完全消失。
自主運動可因肌張力降低,共濟失調或真性肌無力而不能協調,每一動作均突然衝動而出,很不自然,肌力常明顯減弱。嚴重者似癱瘓,稱麻痹性舞蹈症(paralyticchorea)。
妊娠舞蹈病的臨床表現與小舞蹈病相似,多見於17~23歲的初產婦,且多在妊娠有前中期發病,可於妊娠中或分娩后自行停止,亦可於人工流產後立即停止。
(二)慢性進行性舞蹈病(chronicprogressirechorea)又稱Hunt-ington舞蹈病,多於30歲以後起病,是一種常染色體顯性遺傳病,是大腦皮質及紋狀體變性所引起,臨床特點為舞蹈樣動作及進行性疾呆。本病舞蹈樣運動動作慢、幅度大、無節律性,最初癥狀為動作笨拙,不能作精細動作,最後發展成周圍不自主的舞蹈樣動作或舞蹈手足徐動症。舞蹈樣運動在面部表現為面肌張力低,顏面鬆弛,外貌無神狀,表情缺乏,在精神緊張時或談話時面部出現舞蹈樣運動,口半張,撅嘴,舌伸縮亂動,頭不自主前屈、後仰、扭轉或作鬼臉,手指有小的震顫,有時有大幅度運動,以至不能寫字、著衣、進食。行路不穩,常常構音障礙,聲音單調呈鼻音。
(三)老年性舞蹈病(senilechorea)發生於50歲以上的老年人。病理基礎是廣泛散在性軟化灶,尤其是紋狀體部更明顯,臨床表現與Hurtington氏舞病相類似,但發病晚,無遺傳史可作診斷時的參考。
(四)半側舞蹈病(hemichorea)表現為限於一側上下肢的不自主舞蹈運動。它可以是小舞蹈病、慢性進行性舞蹈病的一個部分。亦可以是基底節發生血管性損害的結果。多見於中老年。腦血管病致不完全偏癱時可以出現,舞蹈樣運動可於發後偏癱后立即發生,亦可於數周后或數月後出現,偏癱較完全者,常在偏癱開始恢復后才出現舞蹈動作。這種不自主運動通常以上肢重,下肢和面部輕。
(五)癥狀性舞蹈病(symptomaticchorea)腦部疾病如急性或慢性腦炎、顱內佔位性病變、腦血管疾病、肝豆狀核變性和腦部變性、缺氧、中毒(鉛、汞、鎂、一氧化碳等)、以及全身代謝疾病(如胰島細胞瘤引起的低血糖繼發腦損害)、使用強安定劑等都可發生舞蹈樣運動,稱為癥狀性舞蹈病。
(一)特發性手足徐動症(idiopathicathetosis)病因不明。其特徵是兩側性手足徐動,伴有不同程度的肌張力增高和智能障礙。多為先天性,在生后數月內發病,於顏面、頭部和四肢出現異常運動。智力發育落後,說話較晚或完全不能講話。步行困難,步行時下肢肌張力增高或呈痙攣性,與Little氏病相類似。
(二)癥狀性手足徐動症(symptomaticathetosis)
1.腦性小兒麻痹:在胎兒期或分娩時以及產生造成的大腦損害(早產、妊娠時外傷、分娩是腦缺氧,新生兒重度黃疸等)所致,除有手足徐動癥狀外,尚有各種各處樣的神經、精神癥狀,如在手足徐動側有輕偏癱,錐體束損害征。雙側手足徐動伴有四肢痙攣性不全癱。
2.成人腦卒中後手足徐動:成人腦卒中后出現偏側手足徐動極為罕見,是丘腦症候群的一部分。其它變性疾患(肝豆狀核變性、Schilder-foix氏病,進行性蒼白球萎縮症等)和炎症性疾病(嗜睡性腦炎、百日咳、麻診、白喉等)均可出現手足徐動症。
又名變表性肌張力障礙(dystoniamusculorumdeformans)扭轉性肌張力障礙(torsiondystonia),是軀幹的徐動症。臨床上以肌張力障礙和四肢近端或軀幹順縱軸畸形扭曲為特徵。肌張力在肢扭轉時增高,扭轉停止時則正常。
(一)原發性扭轉性痙攣(primarytorsionspasm)病因不明,常有家族史。主要病理變化為基底節和腦丘腦等部位的神經細胞變性和膠質增生,以尾狀核和殼核改變最明顯。多在5~15歲緩慢起病,最初癥狀大多是一側下肢的輕度運動障礙,足呈內翻跖曲,行走時足跟不能著地,緩慢持續的不自主扭轉性運動以軀幹和肢體近端為最嚴重,引起脊柱前凸和骨盆傾斜,不自主運動累及頸項和肩胛肌時,出現斜頸稱為痙攣性斜頸(spasmodictorticollis)。累及面肌及咽喉時,引起面肌痙攣和構音障礙。智能、肌力和感覺均正常,入睡后不自主運動完全停止。
(二)癥狀性扭轉痙攣(symptomatictorsionspasm)見於感染、血管疾病、中毒(重金屬、藥物、毒氣等中毒)及腫瘤等。
扭轉痙攣的不自主扭轉性運動性質與手足徐動症相同,但前者主要侵犯頸肌、軀幹及四肢的近端肌,而面肌或手肌或全然倖免或僅輕度受累。癥狀性扭轉痙攣可因情緒因素加重,易誤診為癔症。癔症性不自主運動易受暗示影響,而且往往有精神因素為背景。
(一)偏側投擲運動(hemiballismus)為一側肢體猛烈的投擲樣不自主運動,類似舞蹈樣動作,肢體近端重,運動幅度大,力量強,系各種原因引起丘腦底核損害所致,亦見於紋狀體到丘腦底核傳導徑路的病變,如腫瘤、風濕或炎症等。
(二)肌纖維震顫(myokymia)俗稱肉跳。為許多運動單位或一群肌纖維的自發性短暫性搐搦樣收縮。目前認為骨纖維顫搐的產生是因為作用於前角細胞或肌纖維興奮性的營養性影響不一致所致,或是由於運動神經纖維內自發性衝動發放的結果。通常鄰近的肌纖維群交替或間歇發生收縮,表現為肌肉表面的持續波動起伏。此種運動較肌束顫動粗大而持續,但一般並無萎縮。可以是生理性的,見於疲勞、寒冷時,也可見於神經症及體質衰弱的患者或其他代謝性障礙疾病(如大量出血后鹽類丟失,尿毒症)等。在運動神經元疾病時可與肌束顫動一起發生。持續性的單側面肌纖維顫搐可見於腦幹疾病。
(三)肌束顫動和肌纖維顫動(fasciculationsandfibrillations)肌束顫動是指一個肌群或一些肌群細小的快速收縮。為肉眼能見到的最細微的不自主運動。患者亦常講訴有肌肉跳動感。肌纖維顫動是在一塊肌肉的肌腹上數厘米範圍內的細小的快速或蠕動樣的顫動。由於過於細小,肉眼無法看到。肌電圖上表現為小的、反覆的短時間的棘波電位。一般認為肌纖維顫動是脊髓前角細胞或顱神經運動核的刺激現象,而肌束顫動是脊髓前根的刺激現象。
肌束顫動和肌纖維顫動都是下運動神經元損害的重要體征。肌纖維顫動在四肢、顏面、舌肌都可出現,但以舌肌最明顯。這是因為除舌肌外其他部位有皮膚覆蓋不易被發現。伴有肌萎縮的肌束顫動可見於各種惠運動神經元病變,但最常見於變性疾病,如進行性脊肌萎縮症、肌萎縮側索硬化症及進行性延髓麻痹等運動神元疾病。無肌萎縮的肌束顫動亦可見於正常休息時,疲勞與緊張以後,較多見於腓腸肌與手臂肌肉,稱為自發性良性肌束顫動。(四)遲發性運動障礙(tardivedyskinesia)遲發性運動障礙是在長期服用抗精神藥物后出現的不自主動作。最常引起本病的藥物為吩噻嗪類(如氯丙嗉)、和丙其苯基酮類(如氟哌啶醇)。在連續服藥一年後,至少5~10%產生運動障礙。有些病例在服藥3個月後即能罹病。一般認為長期阻斷多巴胺能受體后,後者反應超敏所致。突然停葯可以誘發。由於抗精神病藥物使多巴能神經元長期持續的阻滯,作為適應反應,多巴胺能受體敏感性增高,如果突然停用這些藥物,受體膜的阻滯作用被解除,遞質過多,則會產生或加重遲發性運動障礙。主要表現為:皺額、擠眉弄眼、呶嘴、歪舌、縮著嘴角、舌頭時而伸出,時而縮回、時而左右扭轉、咬牙、反覆張口等,亦有扭轉頸、肩、軀幹、前後屈伸運動、下肢的奇異動作,有時出現聳肩縮背樣動作,急促深呼吸、手指指划運動,有的伴發四肢軀幹的舞蹈樣或手足徐動樣運動,嚴重時出現顫搐、肌陣攣或肌張力增高所致的扭轉性痙攣性運動障礙,痙攣性斜頸等癥狀。癥狀在睡眠中消失,興奮時加重,說話時減輕。異常運動可因步行、運動、叩擊等誘發而加重。
(五)急性肌張力異常(acutedystonias)這是抗精神病藥物所致的急性肌張力障礙,發病機理與突觸間隙內多巴胺有關。常在服用抗精神藥物1~3天內發生,表現為發作性運動增多和肌張力異常,多見於頸、舌、咀嚼肌部位(頸面綜合征),即張口困難、口角歪斜、吐舌、舌有脹滿感和墜落感,牙關緊閉、斜頸或後仰、眼球旋轉或雙眼向上凝視(動眼危象)。嚴重時有角弓反張,下肢過度伸展,扭轉痙攣、肌陣攣等不隨意運動。出現急性肌張力異常,可即刻肌注東莨菪鹼或安定使之緩解。
(六)靜坐不能(akathisia)是抗精神病藥物常見的副反應,可能是腦的非紋狀體部分的DA受體受體阻滯的結果。多發生在接受抗精神病藥物治療的初期,患者表現為無目的的搓手、頓足、反覆徘徊、坐立不安,焦慮苦悶,伴睡眠障礙。因癥狀多在夜間加重,致使患者常在夜間起床於室內來去走動或踏步。藥物所致的靜坐不能癥狀可因停葯而迅速消失,若繼續服藥也會逐漸減輕以至消失。
(七)不寧腿綜合征(restlesssegsyndrome)又稱不寧肢,不寧肢或四肢不安性焦慮,病因不明。一般認為與足部血液循環障礙而致組織代謝產物蓄積有關。部分病例之發病有精神因素或現患有缺鐵性貧血、維生素缺乏、糖尿病等代謝障礙性疾患。主要表現為反覆發作的小腿深部的感覺異常,或自覺似在骨內、狀如蟲爬或奇癢灼熱,難以名狀,伴有惡劣情緒體驗或擴延到大腿及足部,多雙腿對稱性受侵或一側偏重。上肢一般不受累。夜間休息時癥狀加重,活動時減輕,故患者常深夜不寐,輾轉徘徊或移雙腿不已。神經系統無陽性體征,肌電圖正常。
不隨意運動及其病因的診斷與鑒別主要依據病史和神經系統檢查,並結合實驗室檢查及器械檢查。1.病史起病的年齡發生的緩急、癥狀的演變及疾病過程是靜止性還是進行性等特別重要嬰兒期起病者可能是先天性的,應注意其生產史、生長發育史。早產兒及有產傷、窒息史、生后精神及(或)運動系統發育遲緩者提示腦發育不全;產後有嚴重新生兒黃疸者可能為膽紅素腦病引起急性起病者可能為感染(如各種腦炎)或類感染疾病(如風濕熱)。藥物引起者可急性起病(如奮乃靜氯丙嗪類藥物引起的急性肌張力障礙)也可緩慢起病(如吩噻嗪類及丁醯胺類衍生物等神經精神抑製劑所致的遲發性運動障礙)。晚年起病的單側運動過度,即使緩慢起病且病程呈進行性加重也應考慮腦血管病的可能。起病隱襲而呈進行性發展者在任何年齡多提示變性疾病。家族史很重要應該仔細詢問兄弟姐妹及家屬中有無類似或有關疾病,必要時應予檢查。
2.體格檢查神經系統檢查首先應仔細觀察姿勢和體位及不隨意運動的表現,注意其部位、時間(持續的抑或間歇的、速度快慢)幅度(動作的大小几個關節甚至全身)、規律及形態(動作規則刻板抑或多變)。問病史及查體時,都應注意睡眠休息精神及情緒緊張對不隨意運動的影響,錐體外系病變引起的震顫舞蹈樣動作手足徐動扭轉痙攣等,均在精神及情緒緊張時加重睡眠時消失不隨意運動可在肢體遠端明顯,如手足徐動;也可在近端如變形性肌張力障礙可以是緩慢的,產生的異常姿勢可持續數分鐘,如手足徐動及扭轉痙攣;也可很快如舞蹈樣動作手的震顫須觀察上肢在靜止前伸及指鼻試驗等隨意運動時的變化注意肌張力的變化,各種震顫麻痹綜合征的肌張力呈強直性增高,合併有震顫時可呈“齒輪樣強直”。舞蹈樣運動時可有肌張力減低。變動的肌張力是手足徐動症、扭轉痙攣肌張力障礙等的特點。檢查角膜有無Kayser-Fleischer色素環,是確定肝豆狀核變性的簡便而可靠方法。
必要的有選擇性的輔助檢查項目包括:1.腦脊液檢查、血常規、血電解質血糖尿、素氮。
2.疑為肝豆狀核變性的患者應測定肝功能24h尿銅、血清銅、血清銅藍蛋白、血清銅氧化酶活性等以協助診斷。
1.疑有顱內病變可作CT及MRI等檢查;肝豆狀核變性早期行CT檢查可發現基底核已有低密度陰影後期可見廣泛腦萎縮側腦室擴大腦溝增寬;遺傳性舞蹈病患者亦有腦萎縮。2.顱底部攝片胸透心電圖超聲波。
3.對震顫及肌陣攣者可作肌電圖檢查。
相關檢查:尿素、尿銅、腦脊液、銅。
震顫:帕金森氏病、特發性震顫、肝豆狀核變性、多發性硬化、酒精中毒、煤氣中毒、小腦腫瘤、小腦中風等;
舞蹈動作:小舞蹈病(風濕性舞蹈病)、大舞蹈病(遺傳性舞蹈病)、老年舞蹈病等;
手足徐動症:先天性手足徐動症、服用左旋多巴、酚噻嗪等藥物;
其它:抽動穢語綜合征、痙攣性斜頸等。
基本信息
- 中文名
- 不自主運動